






X-linked agammaglobulinemia (XLA), or Bruton agammaglobulinemia, is an inherited immunodeficiency illness because of mutations in the gene coding for Bruton tyrosine kinase (BTK). The illness was first elucidated through Bruton in 1952, for whom the gene is named. BTK is vital to the maturation of pre–B cells to differentiating mature B cells. The BTK gene defect has been mapped to the long arm of the X chromosome at band Xq21.three to Xq22, spanning 37.5kb with 19 exons forming 659 amino acids to complete the BTK cytosolic tyrosine kinase. A database of BTK mutations (BTKbase: Mutation registry for X-linked agammaglobulinemia) lists 544 mutation entries from 471 unrelated households displaying 341 unique molecular situations. No single mutation money owed for greater than 3% of mutations in sufferers. along with mutations, quite a few editions or polymorphisms had been discovered. See the image under.
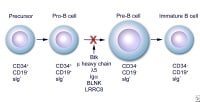 Early stages of B-cell differentiation can be identified by way of the standing of the immunoglobulin genes and with the aid of the cell surface markers CD34, CD19, and floor immunoglobulin (sIg). From: Conley ME. Genes required for B cell building. J Clin invest. 2003;112: 1636-8. Reproduced with permission of american Society for medical Investigation by way of Copyright Clearance middle.
Early stages of B-cell differentiation can be identified by way of the standing of the immunoglobulin genes and with the aid of the cell surface markers CD34, CD19, and floor immunoglobulin (sIg). From: Conley ME. Genes required for B cell building. J Clin invest. 2003;112: 1636-8. Reproduced with permission of american Society for medical Investigation by way of Copyright Clearance middle.The Medscape Reference Pediatrics article, Bruton Agammaglobulinemia, additionally could also be of pastime.
NextPathophysiology
in the absence of BTK, B lymphocytes don't differentiate or mature. with out mature B lymphocytes, antibody-producing plasma cells are also absent. As a end result, the reticuloendothelial and lymphoid organs by which these cells proliferate, differentiate, and are stored are poorly developed. The spleen, the tonsils, the adenoids, the Peyer patches within the intestines, and the peripheral lymph nodes could all be shriveled or absent in individuals with X-linked agammaglobulinemia (XLA).
The protooncogene encoding for BTK has been cloned and its genomic organization decided, allowing an in-depth prognosis of the function of BTK and other signaling molecules in B-cell differentiation.[1]
Mutations in every of the 5 domains of BTK can result in disease. the single most common genetic adventure is a missense mutation. Most mutations result in truncation of the BTK enzyme. These mutations have an effect on very important residues within the cytoplasmic BTK protein and are extremely variable and uniformly dispersed all over the molecule. however, the severity of disease can't be envisioned by the precise mutations. approximately one 1/3 of level mutations affect CGG sites, which on a regular basis code for arginine residues. The putative structural implications of all of the missense mutations are provided in the database.[2, 3, 4, 5]
BTK is necessary for the proliferation and the differentiation of B lymphocytes.[6, 7, 8] males with XLA have a total or almost total absence of B lymphocytes and plasma cells. XLA is an inherited disease that occurs in approximately 1 in 250,000 men. female carriers don't have any clinical manifestations. Infections commence once transferred maternal immunoglobulin G (IgG) antibodies were catabolized, usually at about 6 months of age.
diagnosis
Early detection and prognosis is essential to forestall early morbidity and mortality from systemic and pulmonary infections. The diagnosis is proven through abnormally low or absent numbers of mature B lymphocytes, as well as low or absent expression of the µ heavy chain on the outside of the lymphocyte. Conversely, T-lymphocyte levels are extended. The definitive determinant of XLA is your entire absence of BTK ribonucleic acid (RNA) or protein. particular molecular prognosis is made with the aid of single-strand affirmation polymorphism (SSCP), direct DNA diagnosis, denaturing gradient gel electrophoresis, or reverse transcriptase–polymerase chain response to search for the BTK mutation. SSCP is also used for prenatal evaluation, which may also be carried out by the use of chorionic villus sampling or amniocentesis when a mom is known to be a carrier. IgG ranges less than one hundred mg/dL improve the diagnosis.
hardly ever, the diagnosis is made in adults in their 2nd decade of life. that is regarded as because of a mutation in the protein, somewhat than an entire absence.
PreviousNextEpidemiologyFrequencyUnited States
The estimated frequency of X-linked agammaglobulinemia (XLA) is roughly 1 case per 250,000 population. Two thirds of circumstances are familial, and one 1/3 of cases are believed to come up from new mutations.
world
The incidence of XLA around the world does not fluctuate considerably.
Mortality/Morbidity
Most men with X-linked agammaglobulinemia (XLA) live into their 40s. The prognosis is healthier if treatment is started early, ideally if intravenous immunoglobulin G (IVIG) is began sooner than the individual is aged 5 years. Even with treatment, patients can are expecting to have persistent pulmonary infections, skin disease, inflammatory bowel illness (ulcerative colitis and Crohn illness), and crucial frightened gadget issues as a result of enteroviral an infection.
Race
Most studies involve Northern European sufferers. however, no racial predilection for XLA has been based.
sex
Bruton agammaglobulinemia is an X-linked illness, with best male offspring being affected. Most instances are inherited, however, infrequently, the illness manifests as a result of a spontaneous mutation. Mutations in the gene for the heavy mu gene (IGHM), the immunoglobulin-alpha gene, and the lambda-5 gene can result in agammaglobulinemia, with lower than 1% CD19 expression on B cells. No female carriers present with the clinical manifestations of the BTK mutation.
Age
Male infants change into affected by X-linked agammaglobulinemia (XLA) when maternal antibodies decline regularly after age 4-6 months. If the mother has been identified as a service for the disease, chorionic villi sampling or amniocentesis can be performed to assemble fetal lymphocytes in utero. At birth, wire blood samples may also be examined for a lower in CD19+ B cells and for an increase in mature T cells by the use of fluorocytometric analysis. youngsters most often clinically appear the disease at age 3-9 months with pneumonia, otitis media, cellulitis, meningitis, osteomyelitis, diarrhea, or sepsis. uncommon circumstances of adults in their second decade have been identified with a milder kind XLA regarded as due to a mutation reasonably than an absence of the protein.
PreviousProceed to medical Presentation , Bruton Agammaglobulinemia

No comments:
Post a Comment
Note: Only a member of this blog may post a comment.