![]()
history
Selective immunoglobulin A deficiency (SIgAD) is a primary immunodeficiency disease and is the most typical of the primary antibody deficiencies.[1] complete immunoglobulin A deficiency (IgAD) is defined as an undetectable serum immunoglobulin A (IgA) degree at a value [2, 3]
IgAD is often related to customary B lymphocytes in peripheral blood, normal CD4+ and CD8+ T cells, and, regularly, standard neutrophil and lymphocyte counts. Anti-IgA autoantibodies of the IgG and/or IgE isotype could also be existing. Peripheral blood will also be plagued by autoimmune cytopenias, eg, autoimmune thrombocytopenia,[4, 5] and sufferers may have other autoimmune phenomena.
IgA was first identified by means of Graber and Williams in 1952; ten years later, the first sufferers with IgAD had been described.
IgAD is a heterogeneous dysfunction, and the consequences of intensive study are beginning to elucidate genetic loci and molecular pathogenesis that contribute to quite a lot of subtypes of this dysfunction. several traces of evidence counsel that, in many cases, IgAD and customary variable immunodeficiency (CVID) have a common pathogenesis, which is mentioned additional in Pathophysiology. other data point out totally different genetic chance components. domestic studies exhibit variable inheritance patterns. Familial inheritance of IgAD occurs in approximately 20% of cases,[6] and, within households, IgAD and CVID are associated.[7, 8]
Many IgAD patients are asymptomatic (ie, "customary" blood donors) and are recognized by using finding a laboratory abnormality, with none apparent associated clinical illness. Some patients with IgAD could have the next related conditions: (1) deficits in one or more immunoglobulin G (IgG) subclasses (this bills for 20-30% of IgA-deficient sufferers, many of whom will have whole IgG ranges throughout the commonplace range) or (2) a deficient antibody response to pneumococcal immunization (specific polysaccharide antibody deficiency [SPAD]).
Some sufferers with IgAD later strengthen CVID, and relations of sufferers with CVID could have only selective IgAD. Characterization of the receptor for the transmembrane activator and calcium-modulator and cyclophilin ligand interactor (TACI), encoded by the gene TNFRSF13B ( tumor necrosis factor receptor superfamily member 13B), suggests that individuals with the C104, A181E, and ins204A versions is also at risk for IgAD that progresses to CVID.[9]
primary IgAD is everlasting, and beneath-commonplace levels have been cited to stay static and persist after twenty years of commentary.[10] A recent record paperwork a uncommon case of reversion.[11]
Environmental components equivalent to medication or infections can cause IgAD, but this form is reversible in more than 1/2 the circumstances (see reasons).
even though folks with IgAD have largely been considered healthy, up to date research indicate a higher charge of signs. A 20-12 months follow-up find out about that in comparison 204 healthy blood donors with incidentally identified IgAD to 237 wholesome subjects with standard IgA ranges established that eighty% of IgAD donors and 50% of control topics had episodes of infections, drug allergic reaction, or autoimmune or atopic illness. severe respiratory tract infections passed off in 26% of IgAD subjects, in 24% of subjects with decreased IgA levels, and in eight% of keep an eye on subjects; however, the incidence of lifestyles-threatening infections used to be now not elevated. IgAD is more fashionable in grownup sufferers with continual lung illness than in wholesome age-matched keep an eye on subjects.[12]
sufferers with IgAD are at some increased possibility of growing extreme reactions after receiving blood merchandise.[13, 14, 15] IgG anti-IgA antibodies could lead to severe transfusion reactions if sufferers with IgAD are given complete blood; subsequently, IgA-poor blood or washed crimson cells are most well-liked for those patients. IgA-poor sufferers with immunoglobulin E (IgE)–category anti-IgA antibodies are at risk for anaphylaxis in the event that they obtain blood or intravenous immunoglobulin, but this example is very uncommon. people with such an extraordinary profile will have to receive best low IgA intravenous immunoglobulin preparations. however, caution should be used when administering IGIV to patients with IgAD if their anti-IgA status is unknown.
A history devoid of earlier blood product administration does not exclude the opportunity of anti-IgA antibodies or hostile reactions. fortunately, acceptable precautions can significantly scale back morbidity (see remedy). Blood banks can use a easy ELISA screening option to set up an IgAD blood donor pool.[16]
NextPathophysiology
IgA is the 2nd most typical immunoglobulin in human serum (after IgG) and is the predominant immunoglobulin found in mucosal secretions. Most investigators conclude that extra IgA is in truth produced than every other immunoglobulin, in view that most of it's lost in secretions.
Structurally, IgA has 2 totally different types. Most serum IgA is monomeric, while secretory IgA is a dimer that accommodates an extra protein chain known as the "secretory component;" it's this property that makes this distinctive immunoglobulin proof against the proteolytic enzymes discovered in many human secretions. The IgA dimer and the J-chain that holds the two IgA monomers collectively are produced by B cells. The secretory part is added by way of the serous cells in the mucoserous glands that transport dimeric IgA and other polymeric immunoglobulins (ie, IgM) onto the mucous membranes; a fragment of the receptor/transport protein mediates this translocation. Deficiency of this "polymeric Ig receptor" has been reported, and knock-out mice have been developed in the laboratory. on this state of affairs, the serum IgA level turns into expanded because the IgA can't be transported out of the blood into the secretions.
Secretory IgA antibodies can neutralize viruses, bind toxins, agglutinate bacteria, prevent bacteria from binding to mucosal epithelial cells, and bind to more than a few food antigens, thus fighting their entry into the general circulation. The activities of monomeric serum IgA are usually not totally understood. Dimeric serum IgA most probably represents a kinetically defined pool that has now not yet been transported.
IgAD is a main immunodeficiency illness presumed to outcome from a failure of terminal differentiation in IgA-certain B cells. the improvement of B-lineage cells starts within the fetal liver. B-lineage cell building then transfers to the bone marrow, when it becomes the foremost hematopoietic organ. Pre–B cells become immature immunoglobulin M (IgM)–sure B cells and then migrate from the bone marrow to lymph node germinal facilities. After leaving the bone marrow, the B cells mature and express immunoglobulin D (IgD) receptors, reply to antigens, and, with the help of T cells (CD4+), endure proliferation and class switching and terminal differentiation into plasma cells.[12]
In germinal centers, antigen is offered by follicular dendritic cells with lend a hand from CD4+ T cells and stimulates B cells to proliferate and undergo somatic mutation and immunoglobulin classification-switching. B cells that produce high-antigen affinity antibodies are chosen to develop into plasma cells that produce totally different immunoglobulin isotypes (ie, IgM, IgG, IgA, or IgE) or grow to be recirculating memory B lymphocytes. These techniques are regulated through cell interaction molecules (eg, CD40 on B cells, CD40 ligand on activated T cells, ICOS, TACI-TACIR, and so on), and cytokines (ie, interferon-gamma and interleukin [IL]–2, IL-four, IL-5, IL-6, IL-7, IL-10, IL-12, IL-thirteen, IL-14, and IL-15) and their cell floor receptors.[12]
Most patients with IgAD have an ordinary collection of B cells expressing floor IgA of their blood, however the quantity of surface IgA on each B cell is markedly reduced. according to animal studies, the failure of B cells to terminally differentiate into IgA-secreting plasma cells may be because of the dearth of effects caused by co-stimulatory molecules or cytokines such as IL-4, IL-6, IL-7, or IL-10.
Molecular analyses of B-cell differentiation in a small number of sufferers with selective or partial IgAD recommend that decreased expression degree of alpha germline transcripts earlier than a class change may well be very important for the pathogenesis of some sufferers with SIgAD. A case report from Japan described using reverse transcription polymerase chain reaction (RT-PCR) to separately evaluation alpha1 and alpha2 mRNAs and described a second case of alpha1 gene deletion, which manifested as a partial IgAD.[17] any other that you can imagine clarification is allelic version within the heavy chain 3" regulatory area. a gaggle in Canada evaluated HS1.2 allelic frequencies in 88 SIgAD sufferers and one zero one controls, demonstrating a 39% homozygosity of the allele *1 in patients and 15% homozygosity in controls.[18]
In patients with a partial IgA deficiency, B-cell differentiation may well be disturbed after a class swap.[19] The TACI receptor and 2 ligands (BAFF and APRIL) probably play a task within the pathogenesis of defective humoral immunity. BAFF is B-cell activating issue, and APRIL is a proliferation-inducing ligand. TACI is a tumor necrosis receptor superfamily member that's interested in lymphocyte maturation and survival. In mice, deletion of the sequences coding for TACI or BAFF (B-lymphocytestimulator [BLyS]) intrude with B-cell class switching and end in IgA deficiency.
Missense mutations in one allele of TACI have been found in 4 of 19 unrelated folks with CVID and in 1 of 16 people with SIgAD. The B cells from people with the TACI mutations expressed TACI however did not produce IgG and IgA based on a TACI ligand, a finding notion to replicate impaired isotype switching.[20, 21, 6, 22, 23]
IgAD has been noted to conform into CVID and is often observed in pedigrees that incorporate people with CVID.[24] evidence for a common pathogenesis of CVID and IgAD include shared susceptibility alleles of major histocompatibility advanced classification III genes (D locus),[25] a similar spectrum of IgG subclass deficiencies, a gradual decline of immunoglobulin ranges in concordant siblings, and the development of CVID in some patients with IgAD.
earlier studies of multiple-case families of patients with IgAD confirmed a higher prevalence of CVID among close relatives than within the common inhabitants. In more than one-case households with dominant transmission of CVID and IgAD, CVID was frequently existing in oldsters, followed with the aid of IgAD in the descendants. That study indicated the presence of a predisposing locus within the proximal part of the most important histocompatibility advanced. The recurrence chance used to be discovered to rely upon the sex of the fogeys transmitting the defect. Affected moms were extra prone to produce offspring with IgAD than affected fathers.[8, 26, 27, 7]
IgAD has been stated in sufferers with constitutional chromosome 18 abnormalities, and a case collection of 83 instances of 18p- syndrome confirmed an increased frequency of IgAD; alternatively, attempts to establish a specific locus on chromosome 18 have no longer been successful.[7]
the power of many patients with SIgAD to avoid respiratory infections may relate to compensatory mechanisms at the respiratory mucosal surface and/or compensatory will increase in IgG. Nasal lavage samples received from sufferers with SIgAD compared to customary control show 10-fold higher median IgM ranges and three-fold higher median IgG levels.[28] An extended nasal lavage level of the inflammatory cytokine IL-eight but now not eosinophilic cationic protein (ECP) or TNF-α indicates a degree of subclinical inflammation in these patients.
Nasal biopsy specimens from patients with SIgAD analyzed by way of Brandtzaeg et al related a lower incidence of respiratory tract infections and an extended ratio of IgM-producing cells to IgD-producing cells within the mucosa.[29] patients with recurrent acute rhinosinusitis, otitis media, and tonsillitis had a dominance of the IgD over the IgM isotype, leading the investigators to conclude that immunoregulatory events favoring a dominant local IgD response didn't improve mucosal security. Rose's knowledge confirmed a variety of IgM ranges among patients with IgAD sufferers (zero.87-5.2 μg/mL) however didn't provide correlative clinical knowledge.[28]
Structural lung illness such as persistent obstructive pulmonary disease (COPD) was once up to now idea not to associate having the ability to generate antigen-specific IgA. research of acute exacerbations of chronic bronchitis show that new mucosal IgA to surface-exposed epitopes of the infecting Moraxella catarrhalis isolate developed in sputum supernatants after forty two% of exacerbations,[30] and significant increases in mycoplasmal-specific IgA passed off in 85% of a group of 34 patients hospitalized for acute exacerbations of COPD. In a prospective learn about of 250 hospitalizations for acute exacerbations of COPD, the geometric imply serum titer for IgG and IgA in opposition to Chlamydia pneumoniae used to be larger, with 33% assembly standards for persistent infection.[31] In every other series from India, serum and sputum IgA levels were better in topics with COPD than in control subjects.[32]
recent research, on the other hand, suggest that the mucosal IgA response is impaired in COPD with deficient transport of IgA throughout the bronchial epithelium, presumably involving degradation of the Ig receptor interested in transepithelial routing.[33] Like IgA deficiency, COPD/persistent bronchitis is a heterogeneous dysfunction, and a few cases of major defects in mucosal operate (eg, cystic fibrosis) could in truth be related to increased IgA within the secretions.
Observations that SIgAD is associated with an elevated incidence of atopy suggest a conceivable role for IgA in bronchial asthma pathogenesis. A protecting position of IgA has been seen in murine models of bronchial asthma.[33] It appears likely that in the absence of IgA, mucosal antigen publicity is elevated, which can lead to elevated IgE in opposition to inhalants or meals antigens.
A case regulate find out about evaluated bronchial hyperresponsiveness in children with SIgAD (n = 20), youngsters with commonplace IgA ranges but sensitized to aeroallergens (n = 70) and children with standard IgA ranges and negative skin prick checks (non-atopic) (n = 102). the youngsters with SIgAD had lower pressured vital capacity (FVC) but identical forced expiratory extent in 1 second (FEV1) values. Bronchial hyperreactivity was present in 30-35% of the children in the first 2 groups however in handiest 6% of the keep an eye on crew. The bronchial hyperreactivity among the youngsters with SIgAD correlated with mud mite hypersensitivity however now not with basic atopy.[34]
sufferers with partial IgAD can have diseases through which IgA is relevant to the pathogenesis. for instance, a screening venture recognized 3 instances of partial IgA deficiency in sufferers with dermatitis herpetiformis, with IgA endomysial and tissue transglutaminase antibodies present in 2 of the sufferers. The authors conclude that pathogenically directed IgA antibodies have been sufficient for cutaneous IgA deposition regardless of low serum IgA ranges.[35]
PreviousNextEpidemiologyFrequencyUnited States
At a minimum, an estimated 250,000 folks have IgAD in the us.[36] In African americans, the prevalence of IgAD is 1 case per 6000 individuals. IgA levels are estimated to be abnormally low in 1:500 topics, with the incidence as excessive as 1:a hundred atopic people. full absence of IgA is less general.
international
components associated with the incidence of IgAD embrace a family historical past of IgAD and the u . s . of foundation. family studies using IgAD blood donors as probands exhibit that first-level relatives have a 7.5% prevalence fee of IgAD, which is 38-fold higher than that of unrelated donors.[37] The serological prevalence of IgAD varies one hundred-fold among populations. Prevalences, in lowering order, are as follows:
Arabian peninsula - One in 142 personsSpain - One in one hundred seventy personsEastern Nigeria - One in 255 personsFinland - One in 396 personsCzech Republic - One in 408 personsBasque regions of Spain and France - One in 521 personsCanada - One in 531 individuals[16] Iceland - One in 533 personsEngland - One in 875 personsBrazil - One in 965 personsFrance - One in 3040 personsChina (Han) - One in 2600 personsChina (Zhuang) - One in 5300 personsJapan - One in 14,850-18,500 personsSweden - roughly 20,000 individuals affectedUnited Kingdom - roughly a hundred and twenty,000 persons affected[36] India - None of 3818 blood donors screened; 257 (6.7%) had partial IgAD[38]
isolated IgAD is present in a minority of circumstances of transient hypogammaglobulinemia of infancy. Of a series of forty sufferers offering with recurrent responsive infections, otitis media, bronchitis or bronchial bronchial asthma, or recurrent gastroenteritis when aged four-29 months, only 1 had remoted IgAD, 10 had reduced IgG and IgA ranges, and 6 had diminished IgA and IgM levels.[39] the bulk recovered immunoglobulin levels with the aid of age three years, but three had persistently low IgG and IgA ranges.
A learn about carried out by Weber-Mzell et al on 7293 wholesome white volunteers proven an IgAD occurrence of 0.21% (definition of IgAD was once level [40] the identical study confirmed seasonal fluctuations of serum IgA (SIgA) focus; ranges of SIgA elevated in iciness.
The Latin American crew for primary Immunodeficiency ailments is the usage of a registry strategy to raise dysfunction acceptance in their area. amongst a total of 3321 sufferers registered, fifty three% had an antibody deficiency, of which IgAD was once the more prevalent phenotype.[41]
Mortality/Morbidity
IgAD is extra accepted in adult subjects with power lung illness than in a healthy, age-matched regulate topics.[12]
The 20-yr longitudinal study of wholesome blood donors with incidental findings of IgAD used questionnaires and scientific file evaluations and found a 3-fold elevate in rates of severe childhood respiratory conditions (9% vs 3%), a four-fold raise in rates of extreme adult respiratory prerequisites (sixteen% vs four%), a an identical raise in recurrent delicate respiratory tract infections, and a big increase in charges of recurrent viral infections (16% vs 1%).
This learn about also cited a 4-fold raise within the charge of autoimmune conditions (23% in topics with SIgAD vs 5% in control topics); a 2.5-fold raise within the charge of belly symptoms as a result of milk (sixteen% vs 6%); and moderate increases in the rates of atopic eczema (eight% vs 5%), drug hypersensitivity (9% vs 5%), and meals hypersensitivity (3% vs 1%). A mild decrease used to be observed within the fee of allergic rhinitis and/or eczema (eleven% vs 17%).
When IgAD is related to a number of IgG subclass deficiencies or impaired polysaccharide responsiveness, some people with IgAD could improve recurrent sinopulmonary infections, particularly in patients with concurrent IgG 2 and/or 4 subclass deficiency. This condition has been found to underlie some circumstances of familial deafness as a result of severe recurrent otitis.[42] GI tract infections and issues in sufferers had been stated more continuously with absent secretory IgA, as has an elevated incidence of cancer. Lack of secretory IgA has been hypothesized to compromise the security towards infection with Helicobacter pylori, which is regarded as a result in of abdomen most cancers.
The incidence of most cancers amongst 562 Danish and Swedish subjects with CVID or IgA was once in comparison with that of 2017 loved ones for the duration 1958-1996. among 176 topics with CVID, the incidence of cancer (all sites) used to be elevated (standardized incidence ratio [SIR], 1.8; ninety five% self belief interval [CI], 1-2.9). abdomen most cancers used to be elevated (SIR, 10.3; 95% CI, 2.1-30.2), and malignant lymphoma was once increased (SIR, 12.1; 95% CI, three.3-31). amongst 386 topics with IgAD, the incidence of cancer (all sites) used to be not increased (SIR, 1); then again, the incidence of abdomen cancer used to be elevated, albeit to a mere level (SIR, 5.four; CI, 0.7-19.5).[43] the identical learn about did not show an increase in lymphoid malignancies (non-Hodgkin lymphoma, Hodgkin disease) in IgAD subjects, despite the fact that some proof in the literature signifies that the risk of growing a lymphoid malignancy is increased.[44]
A latest report described 63 Israeli kids with SIgAD adopted for 10 years, with malignancies recognized in three youngsters (four.8%).[45]
sufferers with IgAD who've a compensatory elevate in IgM of their upper respiratory tract secretions and GI fluids are usually less symptomatic. be aware that patients with complete IgAD are on a regular basis more symptomatic than patients partial IgAD.
A prior to now unrecognized clear association of SIgAD with recurrent parotitis of childhood (PTC) was demonstrated by using Fazekas et al in an Austrian pediatric medical institution inhabitants.[46] The occurrence of %in IgA-poor patients (22%) was once a lot greater than in a large inhabitants of wholesome Austrian volunteers (zero.3%).[40] Case studies from different nations toughen this association.[47] Two research provide conflicting evidence of oral conditions in youngsters with SIgAD. A case-managed (n = 34 and n = 111 [control group]) learn about of Hungarian youngsters showed greater decayed, missing, and crammed enamel and enamel surfaces in main dentitions. The severities of mucosal or periodontal disorders have been comparable with the traditional inhabitants.[48] A smaller find out about of Iranian children (eleven circumstances; eleven age- and intercourse-matched controls) confirmed no differences in findings together with dental caries, plaque accumulation and periodontal standing.[49] A extra recent case-keep an eye on study of 32 Icelandic adults with SIgAD when compared with sixty three randomly chosen controls showed an identical ranges of periodontal health and dental well being, but greater rates of tonsillectomy (forty four% vs 24%), adenoidectomy (31% vs eight%), and more pharyngitis, stomatitis, and herpes labialis.[50]
Recurrent sinopulmonary infections are suggested. IgAD regularly manifests as recurrent otitis (in kids), tonsillitis, sinusitis, and bronchitis with extracellular encapsulated bacteria (eg, Haemophilus influenzae, Streptococcus pneumoniae). severe respiratory tract infections occur more often in grownup subjects with IgAD than in normal keep watch over subjects, with a cumulative occurrence fee over 20 years of sixteen%. See the images below.
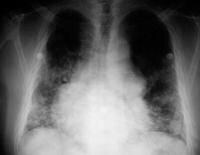
Chest radiograph of a 50-yr-previous man with immunoglobulin A deficiency and extreme bilateral pneumonia. He additionally had congenital coronary heart disease. Serum immunoglobulin G and immunoglobulin M ranges have been commonplace.
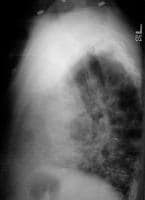
Lateral chest radiograph of a 50-year-old man with immunoglobulin A deficiency and extreme bilateral pneumonia.
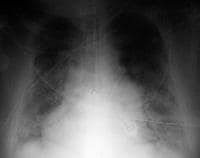
portable chest radiograph of a 50-12 months-previous man with acute respiratory distress syndrome as a complication of severe bilateral pneumonia. The patient died from respiratory failure 2 days after this x-ray film was taken.
The substantial possibility of creating lung injury, which is incessantly stated in patients with CVID, is not a massive possibility to individuals who onlyhave SIgAD. In distinction, lung perform is incessantly significantly impaired amongst patients who have a combination of IgAD and a deficiency of one or more IgG subclasses. a few recently printed circumstances reported the occurrence of hypersensitivity pneumonitis in sufferers with SIgAD and the authors recommend that SIgAD is a chance factor for a more extreme course of the disease and increased susceptibility to improve extrinsic allergic alveolitis.[51, 52]
Autoimmune illness is pronounced in approximately 20% of sufferers with CVID and is also associated with IgAD.
Autoantibodies are regularly produced but could also be tough to observe. The sera of people with IgAD could include quite a lot of autoantibodies that cause no disease or cause myasthenia gravis or thyroid disease. In Sweden, the prevalence of IgAD in thyrotropin-receptor autoantibody–seropositive individuals is 10 instances better than expected within the normal inhabitants.[53] different selective case experiences indicate an association between SIgAD and kind 1 diabetes mellitus, vertigo, vitiligo, and alopecia.Rheumatoid arthritis and systemic lupus erythematosus are the ailments most frequently connected with IgAD.In a survey of serum specimens from 60 healthy subjects with SIgAD, 16 of 21 completely different autoantibody ranges had been larger in IgAD topics than in wholesome regulate topics.[54] A up to date case record described 2 episodes of acute postinfectious glomerulonephritis, separated by means of 15 years, in a 33-yr-outdated man with SIgAD.[55] A northern Italian clinical college described a sequence of 109 patients with autoimmune Addison disease (AAD), 2 of whom had SIgAD. The patients with AAD demonstrated a 12-fold higher prevalence than the local inhabitants.[56]
The occurrence rate of IgG anti-IgA antibodies amongst white persons with IgAD is 30-forty%. In sufferers with blended IgA-IgG subclass 2 deficiency, the speed is 50-60%. In contrast, the occurrence of IgE antibodies towards IgA is very low.
IgA-poor sufferers with anti-IgA antibodies could increase severe reactions when they are transfused with blood elements that incorporate IgA. within the rare cases of actual anaphylaxis, these antibodies are most often of the IgE type; then again, anti-IgA antibodies of the IgG isotype can also result in anaphylactic-kind reactions.[57] even if anaphylactic reactions happen in 1 in 20,000-forty seven,000 transfusions, they represent one of the vital widely wide-spread nonhemolytic causes of transfusion-associated mortality.
A up to date case described a patient with SIgAD who developed an anaphylactic response to a prothrombin complex listen; the authors said that product information didn't list IgAD as a contraindication.[58] A contemporary case report described the successful management of an SIgAD patient who required massive blood transfusion all the way through emergency cesarean delivery.[59] New data quantify the chance of allergies in recipients of blood components containing anti-IgA (ie, blood donated by using SIgAD donors). A Canadian donor database used to be pass-referenced with transfusion response data; the incidence of allergic reactions was once 1.15% within the anti-IgA staff and 2.04% within the group that did not receive anti–IgA-containing blood.[60]
GI tract infections, together with chronic or recurrent giardiasis and other disorders, are suggested with increased frequency. patients with SIgAD have a 10-fold increased possibility of celiac disease. Milk intolerance is popular in patients with main IgAD. reviews indicate that patients with IgAD may have IgG antibodies in opposition to cow milk and ruminant serum proteins. patients with excessive titers of antibodies to cow milk reportedly are more likely to have other autoantibodies.[61] A affected person with SIgAD may still mainfest IgA antibody towards make a choice targets, such as transglutaminase and endomysium, as mentioned in one case of celiac disease by which IgG-particular antibodies have been terrible.[62]
other prerequisites, equivalent to ulcerative colitis, inflammatory bowel disease, Crohn disease, and pernicious anemia, had been described in IgA-poor folks. Friman et al showed that individuals with SIgAD have an elevated possibility of becoming a service of E coli strains that have increased proinflammatory homes, and hypothesize that this will make contributions to the advance of gastrointestinal problems in SIgAD patients.[63] Mucosal infections embody acute diarrhea as a result of viruses, bacteria, or Giardia lamblia parasites. a higher occurrence of serum antibodies to exploit antigens in patients with IgAD suggests that normal serum IgA responses protect the host from continuing exposure to environmental antigens.
An adult sIgAD patient who labored as scaffolder and used to be an avocational aquarist developed concomitant cutaneous Mycobacterium haemophilum and Mycobacterium kansasii infections, infections which are normally viewed handiest in HIV-contaminated or posttransplantation sufferers.[64]
In a case record, a woman with ring chromosome 18 (46XX, r18) had SIgAD in addition to dysmorphic features, failure to thrive, world lengthen of building, hypothyroidism, atopic dermatitis, bilateral continual otitis media and aortic regurgitation with patent foramen ovale.[65]
quality of life (QOL) studies address the cumulative impact of living with a continual disease reminiscent of SIgAD. several standardized scales were administered to all sufferers with main antibody deficiencies considered via a single Norwegian medical institution. Low QOL scores have been associated to unemployment, infections in more than 4 organs, and more than 2 extra ailments. persons with SIgAD had significantly larger QOL index ratings than these with different antibody deficiencies.[66]
Race
IgAD happens in Arab persons at a fee of 1 case per 142 individuals, in white individuals at a rate of 1 case per 500-seven-hundred individuals, in African American individuals at a charge of 1 case per 6000 individuals, and in Asian persons at a price of 1 case per 14,840-18,500 individuals.
sex
A study of 7293 Austrian volunteers showed a greater frequency of SIgAD in males than in girls (0.19% vs zero.014%) and a larger frequency of subnormal serum IgA ranges (zero.07-0.7 g/L) in males (2.sixty six%) than in women (zero.93%).[40]
Age
This disease will also be diagnosed in persons of any age.
reasonable serum IgA levels elevate zero.2 ±0.06 g/L per decade of life.[40]
those older than 6 months who've recurrent higher and lower respiratory tract infections with encapsulated bacteria (eg, H influenzae, S pneumoniae) will have to be evaluated for IgAD as smartly for explicit IgG antibody deficiency, although these could also be maturational delays rather than mounted defects. sufferers with humoral deficiencies don't usually current with recurrent infections in the first few months of existence as a result of they have got circulating IgG from placental transfer. kids and adults may present with recurrent sinopulmonary infections or GI infections or diseases. Case studies exist of severe lifestyles-threatening infections in sufferers with SIgAD.[67, 68, 69] PreviousProceed to medical Presentation , Immunoglobulin A Deficiency







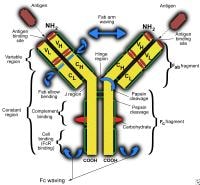 Immunoglobulin G deficiency. Schematic illustration of an immunoglobulin G molecule. CH signifies regular region of heavy chain; CL, consistent area of sunshine chain; VH, variable region of heavy chain; and VL, variable area of sunshine chain.
Immunoglobulin G deficiency. Schematic illustration of an immunoglobulin G molecule. CH signifies regular region of heavy chain; CL, consistent area of sunshine chain; VH, variable region of heavy chain; and VL, variable area of sunshine chain.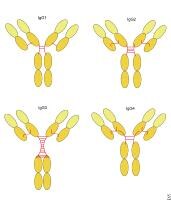 Immunoglobulin G deficiency. Human immunoglobulin G subclasses.
Immunoglobulin G deficiency. Human immunoglobulin G subclasses.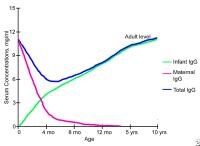 Immunoglobulin G deficiency. modifications in serum immunoglobulin G concentrations throughout infancy and childhood.
Immunoglobulin G deficiency. modifications in serum immunoglobulin G concentrations throughout infancy and childhood.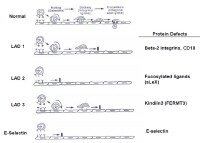 Protein defects. NextPathophysiology
Protein defects. NextPathophysiology