![]()
background
Hypereosinophilic syndrome (HES) is a myeloproliferative disorder (MPD) characterised by continual eosinophilia that's associated with injury to multiple organs.[1, 2, 3, 4, 5, 6] Peripheral eosinophilia with tissue harm has been referred to for approximately 80 years, however Hardy and Anderson first described the particular syndrome in 1968.[7] In 1975, Chusid et al defined the three options required for a analysis of hypereosinophilic syndrome[4] :
A sustained absolute eosinophil depend (AEC) better than >1500/µl is existing, which persists for longer than 6 months.No identifiable etiology for eosinophilia is existing.sufferers will need to have indicators and symptoms of organ involvement.
alternatively, as a result of advances within the diagnostic tactics, secondary reasons of eosinophilia may also be recognized in a share of circumstances that may have otherwise been classified as idiopathic hypereosinophilic syndrome.
The differential analysis (see Differentials and other issues to Be thought to be) of hypereosinophilic syndrome comprises other motives of eosinophilia[1, 8, 9, 10] , which could also be categorized as familial and bought. Familial eosinophilia is an autosomal dominant disorder with a steady eosinophilic count and benign scientific path. got eosinophilia is additional divided into secondary, clonal and idiopathic eosinophilia.[11]
Secondary eosinophilia
Secondary eosinophilia is a cytokine-derived (interleukin-5 [IL-5]) reactive phenomenon. worldwide, parasitic ailments are the most common cause, whereas in developed international locations, allergic diseases are the most common result in.[1] other reasons embrace malignancies (metastatic most cancers, T-cell lymphoma,[12] colon cancer), pulmonary eosinophilia Loffler syndrome,[13] Churg-Strauss syndrome, allergic bronchopulmonary aspergillosis), connective tissue issues (scleroderma, polyarteritis nodosa), skin ailments (dermatitis herpetiformis), inflammatory bowel illness, sarcoidosis, and Addison illness.
Clonal eosinophilia
Clonal eosinophilia is identified by way of bone marrow histology, cytogenetics, and molecular genetics and embody the following:
Acute Leukemia (Pre-B acute lymphoblastic leukemia [ALL], acute myeloid leukemia [AML]-M4EO)chronic myeloid disorders
Molecularly outlined problems embrace the next:
BCR-ABL continual myeloid leukemiaPDGFRA –rearranged eosinophilia (platelet-derived increase issue receptor, alpha polypeptide) (systemic mastocytosis –chronic eosinophilia [SE-CEL])PDGFRβ – rearranged eosinophiliaKIT –mutated systemic mastocytosis
Clinicopathologically assigned issues embody the following:
Myeloproliferative syndrome (MDS)MPDs - traditional MPD (polycythemia) and strange MPD (ie, continual eosinophilic leukemia, systemic mastocytosis, persistent myelomonocytic leukemia)Idiopathic eosinophilia[14, 15, 16, 17, 18, 19]
Idiopathic eosinophilia is a prognosis of exclusion when secondary and clonal causes of eosinophilia are excluded. Hypereosinophilic syndrome is a subset of idiopathic eosinophilia characterized via continual eosinophilia (AEC >1500) of longer than 6 months' period related to organ harm. then again, long-time period follow-up and X-linked clonality studies point out that as a minimum some patients with hypereosinophilic syndrome have an underlying clonal myeloid malignancy or a clonal or phenotypically extraordinary T-cell inhabitants, suggesting a true secondary course of.
assessment of the literature now prefer the view that circumstances of idiopathic HES with FIP1L1 indeed characterize chronic eosinophilic leukemia, as a result of they have molecular genetic abnormality, specifically an FIP1L1–PDGFRA fusion gene.[20] as well as, there are documented instances of acute transformation to both AML or granulocytic sarcoma in some cases of hypereosinophilic syndrome after an interval as long as 24 years. In such instances ,a analysis power eosinophilic leukemia is made in retrospect when acute transformation equipped oblique proof that the condition used to be likely to were a clonal, neoplastic, MPD from the beginning.
in addition, some sufferers with hypereosinophilic syndrome present with options conventional of MPDs, equivalent to hepatosplenomegaly, the presence of leukocyte precursors within the peripheral blood, increased alkaline phosphatase ranking, chromosomal abnormalities, and reticulin fibrosis. Cytogenetic studies in such circumstances is also standard, but molecular genetic research may just show aberrations.
the perfect described aberration is the interstitial deletion on chromosome 4q12, resulting in fusion of the 5’ component to the FIP1L1 gene to the three’ section of the PDGFRA gene. This fusion gene encodes for the FIP1L1–PDGFR alpha protein, the constitutively activated tyrosine kinase process that induces eosinophilia. The incidence of one of these mutation is 0.4% in unselected circumstances of eosinophilia, however it may be as excessive as 12% to 88% in cohorts that meet the arena health group (WHO) standards for idiopathic hypereosinophilic syndrome, specifically those with options of MPD, elevated ranges of tryptase and mast cells within the bone marrow.
sufferers with hypereosinophilic syndrome with the PDGFRA mutation have an awfully excessive incidence of cardiac involvement and raise a bad prognosis with out therapy. luckily, the implications of imatinib treatment in such circumstances of hypereosinophilic syndrome are very encouraging.
the opposite subset of idiopathic eosinophilia, hypereosinophilic syndrome with clonal or immunophenotypically aberrant T-cells, is associated with elevated secretion of IL-5 and cutaneous manifestations. Simon et al stated immunophenotypic abnormality in 16 of 60 sufferers with hypereosinophilic syndrome.[21] furthermore, 9 sufferers had CD3+CD4+CD8- T cells, 3 had CD3+CD4-CD8+ cells, three had CD3+CD4-CD8- cells, and a couple of had CD3-CD4+ cells (1 patient had 2 dissimilar populations). development to T-cell lymphoma used to be seen on this subset of patients with hypereosinophilic syndrome, particularly these with the CD3-CD4+ phenotypes.[21, 22]
continual eosinophilic leukemia[23]
power eosinophilic leukemia is due to independent proliferation of clonal eosinophilic precursors. Simplified standards for the analysis of power eosinophilic leukemia include the next:
Eosinophil count of at the least 1500/µLPeripheral blood blast depend of >2% and a bone marrow blast cell count that's >5% but standards for extraordinary CML, power myelomonocytic leukemia, and continual granulocytic leukemia (BCR-ABL –certain CML) usually are not metMyeloid cells are tested to be clonal (eg, by using detection of clonal cytogenetic abnormality or by demonstration of a very skewed expression of X chromosome genes)
probably the most cytogenetic abnormalities which were described in continual eosinophilic leukemia embody t(5:12) and t(eight:13), and molecular genetic abnormalities include the FIP1L1-PDGFRA fusion gene and ETV6-PDGFRβ.
For very good patient training tools, consult with eMedicine's Cancers and Tumors center. additionally, see eMedicine's patient schooling article Leukemia.
NextPathophysiology
Eosinophil production is governed through a number of cytokines, including IL-3, IL-5, and granulocyte-macrophage colony-stimulating issue (GM-CSF). IL-5 seems to be an important cytokine that's answerable for differentiation of the eosinophil line.[2, 8]
in contrast to neutrophils, eosinophils can survive in the tissues for weeks. Their survival in tissues will depend on the sustained presence of cytokines. simplest eosinophils and basophils and their precursors have receptors for IL-three, IL-5, and GM-CSF. In vitro, eosinophils live on less than 48 hours within the absence of cytokines.
Eosinophil granules incorporate poisonous cationic proteins, which are the principle mediators of tissue harm. These toxins embrace major general protein, eosinophil peroxidase, eosinophil-derived neurotoxin, and eosinophil cationic protein. The latter 2 are ribonucleases. Free radicals produced by means of the eosinophilic peroxidase and the respiratory burst oxidative pathway of the infiltrating eosinophils additional fortify the injury. Eosinophils extend the inflammatory cascade by recruiting extra eosinophils from secreting their very own chemoattractants like eotaxin, platelet-activating factor, and the cytokine RANTES (regulated upon activation, normal T cell expressed, and secreted).
a number of mechanisms were proposed for the pathogenesis of hypereosinophilic syndrome, together with overproduction of eosinophilopoietic cytokines, their superior activity, and defects within the commonplace suppressive legislation of eosinophilopoiesis. Organ damage caused by means of hypereosinophilic syndrome is because of the eosinophilic infiltration of the tissues accompanied with the aid of the mediator liberate from the eosinophil granules. hence, the level of eosinophilia shouldn't be a real reflection of organ injury.
the most severe complication of hypereosinophilic syndrome is cardiac involvement that results in myocardial fibrosis, congestive heart failure (CHF), and loss of life. The mechanisms of cardiac damage should not completely understood, however the harm is marked through severe endocardial fibrotic thickening of either ventricle or both ventricles, resulting in restrictive cardiomyopathy as a result of influx obstruction.
PreviousNextEpidemiologyFrequencyUnited States
various sources indicate that the prevalence of genuine hypereosinophilic syndrome is rare. the commonest cause of eosinophilia in the U.S. is an hypersensitive reaction or allergic illness, however the prevalence of hypereosinophilic syndrome is some distance less.
world
the most typical result in of eosinophilia worldwide is parasitosis. The incidence of hypereosinophilic syndrome is some distance less.
Mortality/Morbidity
Hypereosinophilic syndrome is a power and progressive dysfunction that is doubtlessly deadly. Blast transformation could occur after many years. real idiopathic hypereosinophilic syndrome is typically indolent; on the other hand, patients with traits which are suggestive of a myeloproliferative/neoplastic dysfunction and those who occur CHF have a worse prognosis.
An older evaluate of 57 sufferers with developed hypereosinophilic syndrome stated an average survival of 9 months and a three-yr survival fee of 12%.[4] A later analysis from France cited an 80% survival at 5 years and a forty two% survival at 15 years among 40 sufferers with hypereosinophilic syndrome.[24]
Race
No racial predilection is pronounced for hypereosinophilic syndrome.
sex
there is a male predominance in hypereosinophilic syndrome, with a male-to-female ratio of 9:1.
Age
Hypereosinophilic syndrome is most commonly recognized in sufferers aged 20-50 years, with a top incidence in the 4th decade. Hypereosinophilic syndrome is unusual in youngsters. The incidence of hypereosinophilic syndrome seems to lower within the aged inhabitants.
PreviousProceed to scientific Presentation  Contributor information and DisclosuresAuthor
Venkata Samavedi, MBBS, MD Internist in Houston, TX
Disclosure: Nothing to reveal.
Coauthor(s)
Ronald A Sacher, MB, BCh, MD, FRCPC Professor, interior drugs and Pathology, Director, Hoxworth Blood center, university of Cincinnati tutorial health center
Ronald A Sacher, MB, BCh, MD, FRCPC is a member of the following medical societies: American affiliation for the advancement of Science, American association of Blood Banks, American scientific and Climatological affiliation, American Society for medical Pathology, American Society of Hematology, faculty of yankee Pathologists, international Society of Blood Transfusion, global Society on Thrombosis and Haemostasis, and Royal college of Physicians and Surgeons of Canada
Disclosure: Glaxo Smith Kline Honoraria speaking and instructing; Talecris Honoraria Board membership
Vincent E Herrin, MD associate Professor of medicine, Divisions of Hematology and Oncology, university of Mississippi faculty of drugs
Vincent E Herrin, MD is a member of the next clinical societies: American school of Physicians-American Society of internal medicine and American Society of Hematology
Disclosure: Nothing to expose.
Joe C files, MD Director, Division of Hematology, affiliate Chairman, Professor, division of interior drugs, college of Mississippi clinical middle
Joe C files, MD is a member of the following clinical societies: American affiliation for cancer schooling, American affiliation for the advancement of Science, American faculty of Physicians, American Federation for clinical analysis, American heart affiliation, American scientific association, American Society of Human Genetics, Mississippi State scientific affiliation, ny Academy of Sciences, and Southern scientific association
Disclosure: Nothing to disclose.
Youwen Zhou, MD, PhD, FRCP(C) associate Professor, department of Dermatology and pores and skin Science, university of British Columbia; Director, Hyperhidrosis uniqueness hospital, Co-Director, Psoriasis and Phototherapy Centre, Consulting medical doctor, division of Dermatology, Vancouver normal hospital, Co-Director, Vitiligo and Pigmentation health facility, Oncologist consultant, pores and skin Tumor program, BC cancer agency
Youwen Zhou, MD, PhD, FRCP(C) is a member of the following clinical societies: American Academy of Dermatology
Disclosure: Nothing to disclose.
Paul Schick, MD Emeritus Professor, division of inside medicine, Jefferson scientific faculty of Thomas Jefferson college; analysis Professor, department of inner medication, Drexel university school of medicine; Adjunct Professor of medication, Lankenau sanatorium
Paul Schick, MD is a member of the following medical societies: American college of Physicians, American coronary heart affiliation, American Society of Hematology, world Society on Thrombosis and Haemostasis, and new york Academy of Sciences
Disclosure: Nothing to divulge.
distinctiveness Editor Board
Antoni Ribas, MD Assistant Professor of medicine, Division of Hematology-Oncology, university of California at l. a. clinical center
Disclosure: Nothing to expose.
Francisco Talavera, PharmD, PhD Adjunct Assistant Professor, university of Nebraska scientific middle college of Pharmacy; Editor-in-Chief, Medscape Drug Reference
Disclosure: Medscape earnings Employment
Troy H Guthrie, Jr, MD Director of cancer Institute, Baptist medical center
Troy H Guthrie, Jr, MD is a member of the next medical societies: American Federation for scientific research, American medical affiliation, American Society of Hematology, Florida clinical affiliation, scientific affiliation of Georgia, and Southern clinical affiliation
Disclosure: Nothing to reveal.
Rajalaxmi McKenna, MD, FACP Southwest medical Consultants, SC, department of medicine, just right Samaritan clinic, suggest well being techniques
Rajalaxmi McKenna, MD, FACP is a member of the next scientific societies: American Society of clinical Oncology, American Society of Hematology, and global Society on Thrombosis and Haemostasis
Disclosure: Nothing to disclose.
Chief Editor
Emmanuel C Besa, MD Professor, division of drugs, Division of Hematologic Malignancies, Kimmel cancer center, Jefferson medical college of Thomas Jefferson university
Emmanuel C Besa, MD is a member of the next clinical societies: American affiliation for most cancers schooling, American school of clinical Pharmacology, American Federation for clinical research, American Society of clinical Oncology, American Society of Hematology, and the big apple Academy of Sciences
Disclosure: Nothing to reveal.
References
Seifert M, Gerth J, Gajda M, et al. [Eosinophilia - a challenging differential diagnosis] [German]. Med Klin (Munich). Aug 15 2008;103(8):591-7. [Medline].
Galli SJ, Goetzl EJ. Eosinophils, basophils, and mast cells. In: Handin RI, Stossel TP, Lux SE, Stossel TP, eds. Blood: ideas and practice of Hematology. Baltimore, Md: Lippincott Williams & Wilkins; 1995:621-forty.
Klion advert, Bochner BS, Gleich GJ, et al, and The Hypereosinophilic Syndromes Working workforce. strategies to the treatment of hypereosinophilic syndromes: a workshop abstract file. J allergic reaction Clin Immunol. Jun 2006;117(6):1292-302. [Medline].
Chusid MJ, Dale DC, West BC, Wolff SM. The hypereosinophilic syndrome: prognosis of fourteen instances with assessment of the literature. medication (Baltimore). Jan 1975;fifty four(1):1-27. [Medline].
Simon HU, Rothenberg ME, Bochner BS, Weller PF, Wardlaw AJ, Wechsler ME, et al. Refining the definition of hypereosinophilic syndrome. J allergy Clin Immunol. Jul 2010;126(1):forty five-9. [Medline].
Klion advert. Eosinophilic myeloproliferative issues. Hematology Am Soc Hematol Educ application. 2011;2011:257-sixty three. [Medline].
Hardy WR, Anderson RE. The hypereosinophilic syndromes. Ann Intern Med. Jun 1968;sixty eight(6):1220-9. [Medline].
Wardlaw AJ, Kay AB. Eosinopenia and eosinophilia. In: Beutler E, Lichtman MA, Coller BS, Kipps TJ, eds. Williams Hematology. fifth ed. ny, new york: McGraw- Hill; 1995:844-52.
Tefferi A, Patnaik MM, Pardanani A. Eosinophilia: secondary, clonal and idiopathic. Br J Haematol. Jun 2006;133(5):468-ninety two. [Medline].
Rothenberg ME. Eosinophilia. N Engl J Med. may 28 1998;338(22):1592-600. [Medline].
Gleich GJ, Leiferman KM. The hypereosinophilic syndromes: present ideas and coverings. Br J Haematol. could 2009;a hundred forty five(three):271-85. [Medline].
Helbig G, Wieczorkiewicz A, Dziaczkowska-Suszek J, Majewski M, Kyrcz-Krzemien S. T-cell abnormalities are present at high frequencies in patients with hypereosinophilic syndrome. Haematologica. Sep 2009;94(9):1236-41. [Medline].
Cincin AA, Ozben B, Tanrikulu MA, Baskan O, Agirbasli M. massive apical thrombus in a patient with persistent heart failure and hypereosinophilia: Löffler endocarditis. J Gen Intern Med. Oct 2008;23(10):1713-eight. [Medline].
Bain B. The idiopathic hypereosinophilic syndrome and eosinophilic leukemias. Haematologica. Feb 2004;89(2):133-7. [Medline]. [Full Text].
Bain BJ. Eosinophilia--idiopathic or now not?. N Engl J Med. Oct 7 1999;341(15):1141-3. [Medline].
Oliver JW, Deol I, Morgan DL, Tonk VS. continual eosinophilic leukemia and hypereosinophilic syndromes. notion for classification, literature assessment, and report of a case with a novel chromosomal abnormality. most cancers Genet Cytogenet. Dec 1998;107(2):111-7. [Medline].
Weller PF. The idiopathic hypereosinophilic syndrome. Arch Dermatol. may just 1996;132(5):583-5. [Medline].
Weller PF, Bubley GJ. The idiopathic hypereosinophilic syndrome. Blood. could 15 1994;83(10):2759-79. [Medline]. [Full Text].
Fauci AS, Harley JB, Roberts WC, et al. NIH convention. The idiopathic hypereosinophilic syndrome. clinical, pathophysiologic, and therapeutic concerns. Ann Intern Med. Jul 1982;97(1):78-ninety two. [Medline].
Yamada Y, Sanchez-Aguilera A, Brandt EB, et al. FIP1L1/PDGFRalpha synergizes with SCF to result in systemic mastocytosis in a murine version of persistent eosinophilic leukemia/hypereosinophilic syndrome. Blood. Sep 15 2008;112(6):2500-7. [Medline].
Simon HU, Plötz SG, Dummer R, Blaser okay. atypical clones of T cells producing interleukin-5 in idiopathic eosinophilia. N Engl J Med. Oct 7 1999;341(15):1112-20. [Medline]. [Full Text].
Brugnoni D, Airó P, Rossi G, et al. A case of hypereosinophilic syndrome is related to the enlargement of a CD3-CD4+ T-cell inhabitants able to secrete huge amounts of interleukin-5. Blood. Feb 15 1996;87(four):1416-22. [Medline]. [Full Text].
Bain BJ. Relationship between idiopathic hypereosinophilic syndrome, eosinophilic leukemia, and systemic mastocytosis. Am J Hematol. Sep 2004;seventy seven(1):82-5. [Medline]. [Full Text].
Lefebvre C, Bletry O, Degoulet P, et al. [Prognostic factors of hypereosinophilic syndrome. Study of 40 cases] [French]. Ann Med Interne (Paris). 1989;140(4):253-7. [Medline].
Klion A. Hypereosinophilic syndrome: current solution to analysis and treatment. Annu Rev Med. 2009;60:293-306. [Medline].
Adams JC, Dal-Bianco JP, Kumar G, Callahan MJ. Hypereosinophilic syndrome with attribute left ventricular thrombus proven by contrast echocardiography. Neth heart J. Apr 2009;17(four):169-70. [Medline].
Parrillo JE, Fauci AS, Wolff SM. therapy of the hypereosinophilic syndrome. Ann Intern Med. Aug 1978;89(2):167-seventy two. [Medline].
Luciano L, Catalano L, Sarrantonio C, et al. AlphaIFN-prompted hematologic and cytogenetic remission in power eosinophilic leukemia with t(1;5). Haematologica. Jul 1999;eighty four(7):651-3. [Medline]. [Full Text].
Yamada O, Kitahara okay, Imamura okay, et al. scientific and cytogenetic remission caused through interferon-alpha in a patient with power eosinophilic leukemia associated with a singular t(3;9;5) translocation. Am J Hematol. Jun 1998;58(2):137-41. [Medline]. [Full Text].
Malbrain ML, Van den Bergh H, Zachée P. additional evidence for the clonal nature of the idiopathic hypereosinophilic syndrome: complete haematological and cytogenetic remission precipitated by way of interferon-alpha in a case with a unique chromosomal abnormality. Br J Haematol. Jan 1996;ninety two(1):176-eighty three. [Medline].
Verstovsek S, Tefferi A, Kantarjian H, Manshouri T, Luthra R, Pardanani A, et al. Alemtuzumab remedy for hypereosinophilic syndrome and continual eosinophilic leukemia. Clin most cancers Res. Jan 1 2009;15(1):368-seventy three. [Medline].
Schwartz LB, Sheikh J, Singh A. current strategies in the administration of hypereosinophilic syndrome, including mepolizumab. Curr Med Res Opin. Aug 2010;26(eight):1933-46. [Medline].
Bain BJ. Hypereosinophilia. Curr Opin Hematol. Jan 2000;7(1):21-5. [Medline].
Broustet A, Bernard P, Dachary D, et al. Acute eosinophilic leukemia with a translocation (10p+;11q-). cancer Genet Cytogenet. Apr 15 1986;21(4):327-33. [Medline].
Felice PV, Sawicki J, Anto J. Endomyocardial illness and eosinophilia. Angiology. Nov 1993;forty four(eleven):869-74. [Medline].
Fischkoff SA, Testa JR, Schiffer CA. Acute eosinophilic leukemia with a (10;11) chromosomal translocation. Leukemia. Jun 1988;2(6):394-7. [Medline].
Maubach PA, Bauchinger M, Emmerich B, Rastetter J. Trisomy 7 and 8 in Ph-bad continual eosinophilic leukemia. cancer Genet Cytogenet. Jun 1985;17(2):159-64. [Medline].
Salmon-Nguyen F, Busson M, Daniel M, et al. CALM-AF10 fusion gene in leukemias: simple and inversion-related translocation (10;eleven). most cancers Genet Cytogenet. Oct 15 2000;122(2):137-forty. [Medline].
Schwartz RS. The hypereosinophilic syndrome and the biology of cancer. N Engl J Med. Mar 27 2003;348(thirteen):1199-200. [Medline].
Â

Indurated edematous plaques of hypereosinophilic syndrome on a affected person's legs. Erythroderma in a patient with hypereosinophilic syndrome.
 View desk checklist learn extra about Hypereosinophilic Syndrome on MedscapeRelated Reference themes
Pediatric Hypereosinophilic Syndrome
Dermatologic Manifestations of Hypereosinophilic Syndrome
Pulmonary Eosinophilia
associated news and Articles
Psychological Investigation in sufferers With Polycystic Ovary Syndrome
study Charts Constellation of Turner Syndrome-associated Autoimmune diseases
diminished levels of bodily process in youth With Down Syndrome Are related With Low Bone Mineral Density
Medscape Reference © 2011 WebMD, LLC, Hypereosinophilic Syndrome







 STAT3 gene is diagrammed with depiction of hotspots (areas the place higher numbers of patients were referred to to have mutations). NextPathophysiology
STAT3 gene is diagrammed with depiction of hotspots (areas the place higher numbers of patients were referred to to have mutations). NextPathophysiology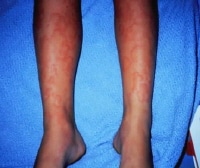 Urticaria associated with a drug reaction.
Urticaria associated with a drug reaction.